Czy wypełnienia amalgamatowe są szkodliwe? Jeśli kiedykolwiek zastanawiałeś się nad zagrożeniami dla zdrowia związanymi z amalgamatem dentystycznym, nie jesteś sam. Istnieje...
Termin ważności i okres przechowywania sterylnych wyrobów medycznych FDA wymaga, aby sterylizowane opakowania narzędzi chirurgicznych/instrumentów posiadały odpowiednią datę...

Jak często szczotkować zęby? Jeśli zastanawiasz się, jak często należy myć zęby, odpowiedź brzmi: dwie minuty, zgodnie z zaleceniami Amerykańskiego Stowarzyszenia...
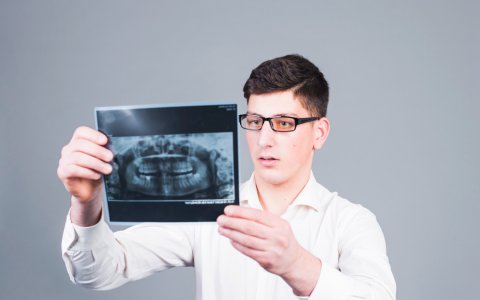
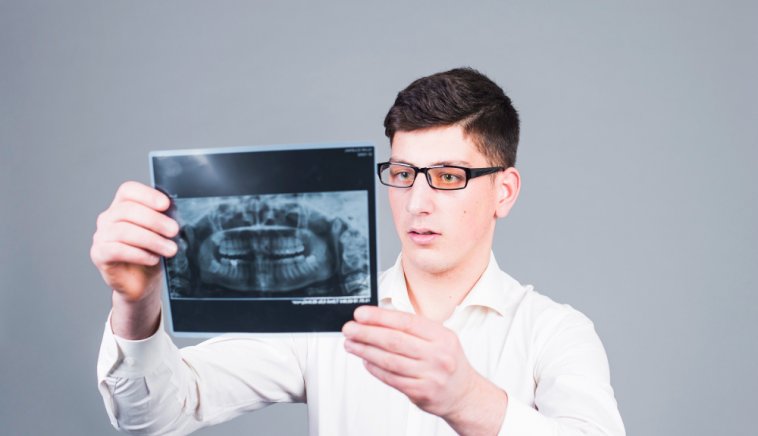
Czym są zdjęcia pantomograficzne? Zdjęcie pantomograficzne zębów może pomóc w określeniu stanu zdrowia jamy ustnej. Są one dobrym sposobem na określenie, czy masz...